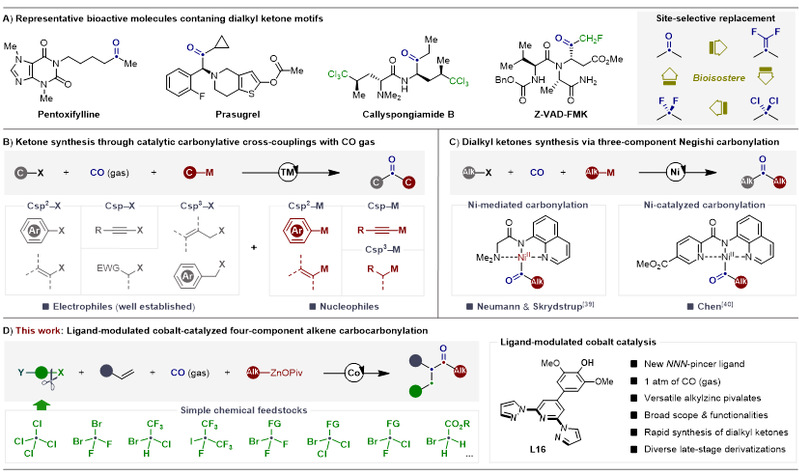
非对称脂肪酮骨架是众多天然产物、生物活性分子及药物中间体中的核心结构单元。羰基不仅可作为药效基团调控药物的药代动力学性质,还可充当二氟亚甲基、二氯亚甲及偕二氟烯基片段的生物电子等排体(图1A)。以CO为C1合成子,基于过渡金属催化直接羰基化是构建羰基类衍生物最为重要的合成工具。然而,该策略下非对称酮的合成研究远远滞后于相应的酯与酰胺合成。已有的羰基化体系高度依赖于贵金属钯催化剂,且其亲电底物范围仍局限于Csp²−X、Csp−X及活化的Csp³−X(图1B)。因此,发展3d金属催化非活化Csp³−X亲电试剂的烷基羰基化高效构建非对称脂肪酮的研究显得尤为重要。近期,Skrydstrup与陈宜峰团队先后报道了基于NNN-钳形镍配合物的三组分Negishi-羰基化偶联策略合成非对称脂肪酮(图1C)。张新刚团队开创性研究也表明,合适的配体可以有效抑制催化惰性Ni(CO)4物种的生成,并能加速镍催化的四组分羰基化偶联反应。相较于镍,廉价金属钴催化的羰基化合成酮类化合物虽然得到了一定的发展,但零星的研究主要局限于对称酮及芳基烷基酮的合成。目前为止,过渡金属钴催化非对称脂肪酮的合成尚未取得突破。基于此,苏州大学李杰课题组设想通过“配体工程"解决钴催化羰基化过程中CO与低价钴配位形成催化惰性的钴羰基物种这一核心难题,从而建立配体调控的钴催化烯烃四组分碳羰基化反应体系,为高效构建非对称脂肪酮提供新的合成范式。
图1. 非对称脂肪酮的研究现状。
图片来源:J. Am. Chem. Soc.
近年来,苏州大学李杰课题组长期致力于新型金属有机试剂的设计与制备,拓展其在过渡金属催化交叉偶联反应体系中的可控转化等方面的研究,发展了基于“抗衡阴离子效应”调控有机锌试剂稳定性与反应性的研究策略(Nat. Commun., 2021, 11, 4366; Angew. Chem. Int. Ed., 2022, 61, e202202379; Nat. Commun., 2023, 14, 1454-1465; Chem. Sci., 2023, 14, 8672; ACS Catal., 2023, 13, 9254; Chem. Sci., 2024, 15, 16250; Angew. Chem. Int. Ed., 2021, 60, 15497; J. Am. Chem. Soc., 2019, 141, 98; J. Am. Chem. Soc., 2019, 141, 18127; Angew. Chem. Int. Ed., 2024, 63, e202408211; Sci. Bull., 2024, 69, 3334; Angew. Chem. Int. Ed., 2025, 64, e202421190; J. Am. Chem. Soc., 2025, 147, 32238; Nat. Commun., 2025, 16, 8803; Natl. Sci. Rev., 2026, 13, nwag011; J. Am. Chem. Soc., 2026, DOI: 10.1021/jacs.6c05845)。为解决上述挑战,本研究开发一种可精准调控钴催化烯烃四组分碳羰基化反应的新型双吡唑吡啶型NNN-钳形配体,在1 atm CO氛围下实现了活化与非活化烯烃、烷基锌试剂和烷基卤化物之间的高选择性羰基化偶联,解锁了非对称烷基酮,尤其是传统方法难以合成的非对称烷基酮的高效合成(图1D)。
首先,作者以廉价的简单多卤代小分子CCl4(1a)、乙基特戊酸锌试剂(3a)和茚(2a)为模板底物,以CoI₂为催化剂,1,4-二氧六环为溶剂,在常压一氧化碳(CO)下进行四组分羰基化偶联反应的条件探索。当采用联吡啶、菲啰啉或三联吡啶等常见双氮或三氮配体骨架时,目标产物的收率仅为8%–16%。酰胺型三齿氮配体L5虽曾成功用于镍催化的三组分羰基化Negishi交叉偶联反应,但在本体系中仅获得14%的收率。值得注意的是,使用结构更加刚性的NNN-钳形配体L6‒L10后,产物收率显著提高至65%。在此基础上,进一步以2,6-双(N-吡唑基)吡啶配体(bpp,L10)为骨架,考察了吡啶环上不同取代基对反应的影响(L12‒L16)。其中,吡啶环C4位含有供电子基团(4-羟基-3,5-二甲氧基苯基)的配体L16表现最佳,在2小时内以78%的收率得到非对称脂肪族酮4(图2a)。随后,以L13与L16为代表性三齿钳形配体,对不同钴配合物及反应溶剂进行了筛选,最终确定CoI₂为最佳催化剂,1,4-二氧六环为最优反应溶剂(图2b、2c)。此外,制备了包括传统卤素配位烷基锌试剂在内的6种亲核试剂,并开展了一系列平行实验,以探究阴离子配位调控策略对烷基锌试剂反应性的影响。结果表明,与其它阴离子配位的烷基锌试剂相比,特戊酸(OPiv)配位的锌试剂在该反应体系中表现出最优的反应活性与选择性(图2d)。
图2. 条件优化及烷基锌试剂底物范围。
图片来源:J. Am. Chem. Soc.
随后,以CCl₄为自由基源,系统考察了烷基锌试剂在钴催化烯烃碳羰基化反应中的底物适用范围(图2e)。结果表明,该催化体系兼容芳基醚、烯烃、三氟甲基、烷基氯、酯基及缩醛等多种官能团。值得一提的是,三元至六元环状烷基锌试剂均能顺利参与反应,以45%‒83%的收率得到双环甲酮类化合物15‒19。其中,化合物19的结构由单晶X射线衍射分析进一步证实。值得注意的是,该反应体系还可拓展至支链二级烷基锌试剂,以中等收率快速构建结构复杂的非对称二烷基酮20‒21。此外,所得产物可通过基于双重C–Cl键断裂的分子编辑策略,进一步转化为萘骨架化合物22–24(图2e,底部),从而充分证明了该产物的合成应用价值。接着,又以CF₂Br₂为自由基前体,考察了芳基烯烃在该多组分羰基化反应中的适用范围(图3a)。该反应对芳基烯烃的电子效应及空间位阻不敏感,并能兼容酯基、吗啉基、氰基及吲哚基等多种官能团。对于含有芳基卤化物、苄氯或烷基溴的底物,钳形钴催化剂表现出优异的化学选择性,优先在双键上发生二氟甲基化羰基化反应,且未观察到Negishi偶联副产物。多种脂肪族烯烃同样能高效参与反应,生成相应的二烷基酮(图3b)。该体系可兼容含有Csp³–Cl、Csp³–Br、Csp³–I以及芳基Csp²–Br、Csp²–Cl键的脂肪族烯烃。此外,含有三氯甲基、三氟甲基、酯基、砜基、烷基氯、溴及全氟烷基等官能团的烷基卤化物,均可通过选择性断裂Csp³–X(X = Cl, Br, I)键,高效构建非对称脂肪酮化合物。
图3. 烯烃和烷基亲电试剂底物范围。
图片来源:J. Am. Chem. Soc.
此外,该四组分羰基化反应成功将偕二氟亚甲基和羰基片段引入至卡格列净、非布司他、水杨苷及吡丙醚等药物分子中,以50%‒70%的收率得到高附加值的氟代脂肪族酮。值得注意的是,该四组分偶联反应可以顺利放大至克级规模并保持反应效率不变。以化合物80为模型底物,通过Wittig环化、格氏反应及串联还原环化等策略实现了碳骨架的快速编辑,以中等至良好收率得到化合物81‒83。此外,从化合物84出发,成功构建了含偕二氟亚甲基的新型己酮可可碱类似物87。上述应用充分彰显了该方法在药物发现中高效、多样化制备高官能团化分子的通用性(图4)。
图4. 生物活性分子的修饰及后期合成应用。
图片来源:J. Am. Chem. Soc.
随后,作者开展了一系列机理探究实验以阐明该反应机制。自由基捕获实验及自由基钟实验表明,该羰基化反应涉及自由基接力过程(图5a、5b)。进一步的对照实验提供了关键佐证:在无烷基锌试剂参与时,ATRA过程未能发生,底物90被回收;而使用1.0当量芳基特戊酸锌还原原位生成的CoI物种时,则可检测到二氟甲基化羰基化产物93。上述结果表明,有机锌试剂对于原位生成具有催化活性的低价钴物种至关重要,该物种进而引发自由基接力羰基化偶联反应(图5c)。
图5. 机理研究及可能的催化循环。
图片来源:J. Am. Chem. Soc.
为阐明钴配合物在羰基化反应中的作用,作者合成了钳形钴配合物94,并利用单晶X射线衍射确认其结构(图5d,上图)。实验表明,该CoII配合物可直接催化烯烃羰基化反应,以66%产率得到产物36。为验证CO插入过程,作者设计了串联实验:首先,在配合物94存在下,于1 atm CO中对烷基锌试剂3k进行羰基化;随后,将该反应混合物转移至含CF₂Br₂和2i的N₂气氛体系中,以28%产率得到36(图5d,中图)。该结果证实配合物94可介导CO与烷基锌试剂的1,1-插入反应,形成酰基钴中间体。此外,配合物94经1.0当量对甲基苯基特戊酸锌试剂还原30分钟生成的低价钴物种,可直接驱动目标多组分羰基化反应,以63%产率高选择性地得到非对称二烷基酮36(图5d,下图)。基于上述实验结果,新生成的低价钴物种可能为该反应的催化活性物种。
因此,基于上述机理研究以及前人的工作,作者提出了一种可能的催化循环机理。首先是CoII配合物被烷基锌试剂还原,生成活性CoI物种A。随后,A与烷基锌发生转金属化生成烷基-CoI物种B,B与CO气体发生1,1-插入反应成酰基-CoI中间体C。烷基卤化物1与中间体C发生分子间卤素原子转移(HAT)过程,生成酰基-CoII中间体D,同时释放出Csp3自由基E。E进一步与烯烃2发生自由基加成,生成相应的仲烷基自由基F。F再与酰基-CoII中间体E发生自由基加成生成关键的CoIII中间体G。最后,G经还原消除得到非对称二烷基酮目标产物,并再生CoI配合物A(图5e)。此外,亦存在另一条可能的反应途径,即涉及中间体D与自由基F之间的自由基取代过程。
总结
综上所述,李杰教授团队发展了一种基于自主设计的新型双吡唑基吡啶型NNN-钳形配体调控的钴催化四组分碳羰基化反应。该反应实现了活化及非活化烯烃、烷基锌试剂、烷基卤化物与一氧化碳之间的高选择性偶联,在1 atm CO条件下高效构建非对称脂肪酮,解决了传统方法难以合成非对称脂肪酮的难题。实验结果表明,该配体显著调控了钴中心的反应性,既避免低价钴与CO形成失活羰基络合物,又有效促进自由基接力型羰基化过程。该方法具有操作简便、官能团兼容性广、区域选择性与化学选择性优异等显著特点,为直接合成结构不对称的脂肪族酮类化合物开辟了一条新途径。
相关研究发表在Journal of the American Chemical Society上,苏州大学研究生曾婷和阮良捷为论文的共同第一作者,苏州大学李杰教授、中国科学院兰州化学物理研究所何林研究员为共同通讯作者,苏州大学为第一通讯单位,该研究得到了国家自然科学基金、江苏省自然科学基金、以及苏州市科技计划项目的经费支持。
原文(扫描或长按二维码,识别后直达原文页面):
Ligand-Modulated Cobalt-Catalyzed Alkene Carbocarbonylation Unlocks Aliphatic Ketone Synthesis
Ting Zeng, Liangjie Ruan, Zhili Cui, Jixin Wang, Changrui Nie, Shulei Ge, Lin He*, Jie Li*
J. Am. Chem. Soc., 2026, DOI: 10.1021/jacs.6c06343
研究团队介绍
李杰,苏州大学材料与化学化工学部教授。2015年于德国哥廷根大学取得博士学位,2017年至2019年在德国慕尼黑大学化学院从事博士后研究工作,2020年1月起就职于苏州大学。研究领域包括1. 设计、制备新型金属有机试剂;2. 基于新试剂发展过新型偶联反应。
https://www.x-mol.com/groups/JieLi